La troponina I como marcador de daño miocárdico de tipo isquémico. A propósito de un caso clínico
Resumen breve
La troponina I cardíaca (TnIc) es un biomarcador muy sensible y específico de lesión miocárdica1. Sus usos más frecuentes en medicina veterinaria son: identificación de patologías cardíacas en fase preclínica, valoración del impacto de patologías sistémicas a nivel cardíaco, discriminación entre causas no cardíacas o cardíacas en pacientes con distrés respiratorio y como factor pronóstico2.…Índice de contenidos
Resumen
La troponina I cardíaca (TnIc) es un biomarcador muy sensible y específico de lesión miocárdica1. Sus usos más frecuentes en medicina veterinaria son: identificación de patologías cardíacas en fase preclínica, valoración del impacto de patologías sistémicas a nivel cardíaco, discriminación entre causas no cardíacas o cardíacas en pacientes con distrés respiratorio y como factor pronóstico2. En este artículo exponemos el caso de un paciente con una grave patología sistémica y alteraciones cardíacas en el que la medición de la TnIc fue de gran ayuda para poner de relieve la presencia de un daño miocárdico isquémico grave y para monitorizar la patología a lo largo del tiempo. En el texto se describen las técnicas diagnósticas empleadas, el protocolo terapéutico y la evolución del paciente.
Introducción
La troponina es una proteína que forma parte del aparato contráctil de las células musculares3. La unidad contráctil del músculo estriado está constituida por diferentes filamentos proteicos gruesos y finos que se agrupan de forma organizada en una unidad estructural llamada miofibrilla3. Los filamentos gruesos están compuestos principalmente por abundantes polímeros de miosina y otras proteínas citoesqueléticas como la titina, la proteína C de unión a la miosina o la proteína M. Los filamentos finos están constituidos por polímeros de actina que además incluyen otras proteínas como la tropomiosina y las tres proteínas del complejo troponina3.
El fenómeno de relajación-contracción de las células musculares ocurre cuando se forman o deshacen uniones entre los filamentos de actina y miosina. Los elementos fundamentales para la formación del complejo actina-miosina son: la actividad ATPasa de las cabezas de miosina, el complejo troponina-tropomiosina de la actina y la presencia de iones calcio y ATP3. El complejo troponina está formado por tres subunidades:
- troponina I (TnI): cuya función es inhibir la interacción entre la actina y la miosina.
- troponina T (TnT): une el complejo troponina a la tropomiosina
- troponina C (TnC): contiene el punto donde se liga el calcio necesario para el fenómeno de contracción-relajación.
La contracción se desencadena por el aumento del calcio citosólico, proveniente tanto del espacio extracelular como del retículo sarcoplásmico, durante la fase 2 del potencial de acción. El calcio se une a la troponina C que provoca un cambio en la conformación del complejo troponina-tropomiosina con la consecuente exposición de los sitios activos de los filamentos de actina. Este cambio de conformación permite que las cabezas de miosina interactúen con la actina y formen los llamados puentes o enlaces cruzados. La energía necesaria para producir la contracción muscular es proporcionada por el ATP. La secuencia de contracción y relajación puede resumirse en 4 fases: dos en las que la actina y la miosina se separan (estado relajado y estado relajado energizado) y dos en las que la miosina está ligada a la actina (complejos activos y complejo de rigor)3.
La liberación de la troponina al torrente circulatorio puede ocurrir tras un daño reversible o irreversible de la célula cardíaca (tabla 1) y numerosas patologías cardíacas, y no cardíacas, pueden estar implicadas en el proceso (tabla 2)1,4. La troponina es un marcador específico de daño miocárdico, pero no específico de la causa que lo provoca. El grado del daño, y por tanto la cantidad de troponina liberada, estará íntimamente relacionado con la gravedad del insulto. En medicina humana, la troponina es uno de los principales marcadores biológicos de necrosis miocárdica en el síndrome coronario agudo5. Las 3 subunidades de la troponina son comunes tanto al músculo cardíaco como al músculo esquelético, pero la TnI y la TnT presentan isoformas específicas para cada tejido (la TnC presenta la misma isoforma tanto para el tejido cardíaco como el esquelético)3.
| Tabla 1. Mecanismos responsables de la liberación de troponina desde la célula cardíaca. |
| Irreversibles | Necrosis | Daño isquémico, inflamatorio, tóxico o traumatismo |
| Apoptosis | Muerte celular programada | |
| Renovación celular cardíacas | Regeneración natural de las células cardíacas | |
| Reversibles | Aumento de la permeabilidad | Aumento del estrés celular como consecuencia del trabajo miocárdiaco. Puede acompañarse de daño isquémico |
| Proteolisis intracelular | Liberación de los productos de degradación de la troponina | |
| Formación de vesículas | Liberación de troponina y sus productos de degradación a través de formación de vesículas en las membranas | |
| Tabla 2. Patologías cardíacas y no cardíacas asociadas a daño miocárdico más frecuentes en el perro. |
| Cardíacas |
|
| No cardíacas |
|
Esta característica hace que la TnI y TnT cardíacas (TnIc y TnTc) sean utilizadas como biomarcadores específicos de daño miocárdico. Una pequeña cantidad de TnIc y TnTc está disuelta en el citoplasma de los cardiomiocitos y la mayor parte se encuentra ligada al complejo troponina-tropomiosina5. Esta distribución de las proteínas en la célula define en parte el patrón de liberación de la troponina al torrente sanguíneo tras el daño de la célula cardíaca. Generalmente se produce primero una liberación rápida de troponina desde el citosol que es seguida de una liberación más lenta desde la zona estructural5.
Los niveles séricos de troponina son variables y dependen tanto de la cantidad que se libera desde la célula cardíaca, íntimamente relacionada con la etiología de proceso, como la tasa de eliminación (renal y sistema retículo-endotelial)1. Por este motivo no existe una definición clara de la variación de los niveles de troponina a lo largo del tiempo ni de unos límites de referencia según la gravedad de la patología. En líneas generales, después del daño cardíaco puede observarse un aumento de los niveles séricos de troponina en las primeras 2-3 horas alcanzando un pico a las 18-24 horas1.
Los niveles séricos de TnIc o TnTc son prácticamente inapreciables en animales sanos. Los valores mínimos en cada paciente suelen estar asociados al recambio celular normal y las variaciones biológicas del propio individuo o de la especie. Los métodos analíticos validados en medicina veterinaria son aquellos que determinan los valores de TnIc.
Los valores de normalidad descritos en medicina veterinaria son numerosos y probablemente no comparables entre ellos porque han sido realizados con técnicas de laboratorio diferentes, pero se considera normales valores de TnIc inferiores a 0,1- 0,2 ng/ml tanto en el gato como en el perro1. Las concentraciones de troponina en patologías cardíacas (congénitas o adquiridas) suelen ser inferiores a 1 ng/ml y no suelen ser mayores (1-2 ng/ml) a menos que se compliquen, por ejemplo, con un cuadro de insuficiencia cardíaca congestiva1.
Como se ha descrito previamente en medicina humana, el daño miocárdico más grave suele estar asociado a las cardiopatías isquémicas (ej. infarto miocárdico) o las cardiomiopatías inflamatorias (miocarditis) y, por tanto, cursan con una mayor elevación de los niveles de TnIc (> 10 ng/ml)5. Los pacientes con patologías sistémicas suelen tener un grado variable de daño miocárdico que se manifiesta con valores de TnIc generalmente inferiores a 1 ng/ml, pero los pacientes críticos pueden tener un daño miocárdico mayor y los valores de TnIc pueden superar los 10 ng/ml1. En medicina veterinaria la TncI es un marcador biológico de gran utilidad como factor pronóstico en pacientes que presentan patologías sistémicas graves (ej. dilatación-torsión gástrica, hemoabdomen, síndrome de respuesta inflamatoria sistémica)6-8. El aumento de la TncI en estos pacientes está asociada a un pronóstico más desfavorable6-8
Caso clínico
Se describe el caso clínico de una hembra esterilizada, raza Labrador, de 10 años y 35 Kg de peso. Fue remitida al servicio de hospitalización de nuestro centro para su estabilización, tras haber realizado una esplenectomía de urgencia en su centro veterinario. La causa de la intervención fue un hemoabdomen agudo asociado a una masa esplénica con sangrado activo. El procedimiento quirúrgico, la anestesia y el período postoperatorio del paciente transcurrió sin complicaciones hasta que se observó, en la monitorización electrocardiográfica, la aparición de arritmias ventriculares, momento en el cual fue remitida a nuestro centro.
Cuando el paciente ingresó en el hospital se realizó una exploración física general en la que se observó que el paciente estaba alerta. Las mucosas estaban ligeramente pálidas y con un tiempo de relleno capilar inferior a 2 segundos. Su frecuencia respiratoria era de 60 respiraciones por minuto y la frecuencia cardíaca de 180 latidos por minuto. Se monitorizó la presión arterial sistólica, diastólica y media (PAS, PAD y PAM) mediante método oscilométrico de alta definición (B.Braun®). La PAM presentaba un valor entre 75-90 mmHg. Mostraba una marcada resistencia a la palpación abdominal, el pulso era débil y su temperatura rectal era 38.2ºC. Los signos clínicos del paciente eran compatibles con un shock hipovolémico en fase de compensación temprana debido a la hemorragia interna abdominal previamente descrita.
Las alteraciones observadas en la analítica sanguínea prequirúrgica de su veterinario fueron anemia leve (27.8%, valor de referencia 37.3-61.7%) y leucocitosis (21.99x10^9/L, valor de referencia 5.5-16.9x10^9/L) con desviación a la izquierda y moderada trombocitopenia (76x10^9/L, valor de referencia 100-518x10^9) (Tabla 3). La anemia era normocítica-normocrómica, siendo el diagnóstico diferencial más probable una anemia hemorrágica pre-regenerativa debido al sangrado abdominal (los reticulocitos tardan en aparecer en el torrente sanguíneo de 2 a 3 días desde el inicio de la hemorragia). Sin embargo, no se podía excluir una anemia no regenerativa como consecuencia de una enfermedad inflamatoria crónica.
| Tabla 3. Hemograma prequirúrgico . |
| Parámetro | Valor | Intervalo de referencia |
| Leucocitos | 21.99 x 10ˆ9/L | 5.5-16.9 |
| Neutrófilos | 18.07 x 10ˆ9/L | 3.12-11.6 |
| Linfocitos | 2.27 x 10ˆ9/L | 0.73-7.86 |
| Monocitos | 1.62 x 10ˆ9/L | 0.07 – 1.36 |
| Eosinófilos | 0.02 x 10ˆ9/L | 0.06 – 1.93 |
| Hematocrito | 27.8% | 37.3 – 61.7 |
| Eritrocitos | 4.21 x 10ˆ9/L | 4.60 – 10.20 |
| MCV | 66,0 fL | 81.6 – 73.5 |
| MCH | 24,0 pg | 21.2 – 25.9 |
| Hemoglobina | 10.1 g/dL | 12 - 18 |
| Plaquetas | 76 x 10ˆ9/L | 100 - 518 |
| Basófilos (%) | 21.99 x 10ˆ9/L | 0-1,30 |
| Monocitos (%) | 21.99 x 10ˆ9/L | 0,70-4,70 |
La leucocitosis neutrofílica con desviación a la izquierda, característica de un aumento agudo de neutrófilos inmaduros, podía ser compatible con un proceso inflamatorio/infeccioso de carácter agudo. La trombocitopenia moderada era probablemente secundaria a un excesivo consumo como consecuencia del sangrado o una coagulación intravascular diseminada. La bioquímica reveló una leve azotemia (Tabla 4) probablemente de origen prerrenal por la disminución de la perfusión renal como consecuencia de la hipovolemia, sin embargo, no se realizaron pruebas complementarias para clasificar el daño renal del paciente.
| Tabla 4. Bioquímica prequirúrgica. |
| Parámetro | Valor | Intervalo de referencia |
| Glucosa | 118 mg/dL | 70-125 mg/dL |
| BUN | 40 mg/dL | 7-27 mg/dL |
| Creatinina | 2,4 mg/dL | 0.3-1.4 mg/dL |
| PT | 5 g/dL | 5.5-7.8 g/dL |
| Albúmina | 2.3 g/dL | 2.2-3.9 g/dL |
| ALP | 125 U/L | 25-190 U/L |
| ALT | 67 U/L | 9-60 U/L |
| Globulinas | 2.7 g/dL | 2.5-4.5 g/dL |
En el momento de la llegada del paciente a nuestro centro realizamos un hematocrito manual que dio como resultado un 22%. La leve reducción del hematocrito probablemente era secundaria a las pérdidas de sangre que se produjeron durante la esplenectomía. A pesar de que la reducción era leve, cabe recordar que los perros en shock hipovolémico en fases tempranas de compensación son capaces de liberar hasta un 35% de la volemia por contracción esplénica, lo que podría llevar a infravalorar la gravedad de la hemorragia del paciente en un primer momento. Se realizó una ecofast abdominal donde se descartó la presencia de líquido libre peritoneal, excluyendo de esta manera la recidiva del hemoperitoneo.
La medición del lactato reveló una moderada acidosis láctica (4,3 mmol/L, ref: <2,5 mmol/L) probablemente como resultado de una hipoperfusión sistémica e hipoxia tisular durante el sangrado (hipovolemia severa y anemia). Se realizó un electrocardiograma de superficie que mostró un ritmo sinusal interrumpido por escasos complejos ventriculares prematuros polifocales aislados.
Como estas alteraciones electrocardiográficas no suponían ningún impacto hemodinámico en el paciente en el momento del estudio no fue necesario iniciar un tratamiento antiarrítmico. Se hospitalizó con monitorización continua de sus constantes vitales y con un tratamiento médico intravenoso basado en antibioterapia de amplio espectro (amoxicilina-ácido clavulánico 22 mg/kg/IV cada 8 horas) y analgesia a base de lidocaína (3mg/kg/h) y ketamina (0,15 mg/kg/h) en infusión continua, tramadol (3 mg/kg/IV cada 8 horas) y paracetamol (10 mg/kg/IV cada 8 horas). Además, se realizó una fluidoterapia agresiva para la recuperación del volumen circulante. Se administraron cristaloides (solución polielectrolítica balanceada, ringer lactato) con un bolo inicial de 15 ml/kg en 20 minutos hasta conseguir un aumento de la PAM de 90 mmHg. Luego se continuó con una fluidoterapia de mantenimiento en función de estado del paciente.
Transcurridas 24 horas del ingreso hospitalario se observó un empeoramiento del cuadro arrítmico. El electrocardiograma de superficie mostraba salvas de ritmo idioventricular acelerado polimorfo alternadas con períodos de taquicardia ventricular monomorfa con morfología de bloqueo de rama izquierda (figura 1). Como consecuencia de las condiciones clínicas no fue posible realizar un estudio ecocardiográfico completo y se completó el examen cardiológico con una ecocardiografía fast. Los hallazgos ecográficos evidenciaban una leve reducción de la función sistólica biventricular más acentuada en la contracción radial del ventrículo derecho.
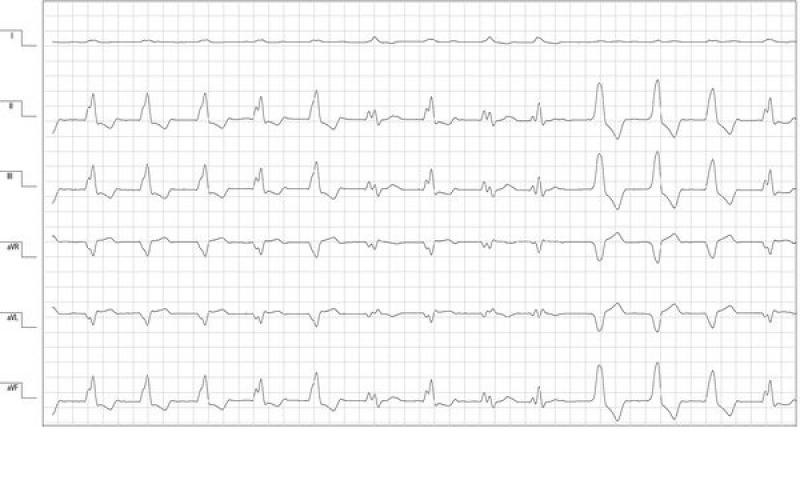
Las alteraciones electrocardiográficas y ecográficas eran sugerentes de un daño miocárdico. Vista las alteraciones hemodinámicas y metabólicas en curso, y la ausencia de antecedentes de patologías cardíacas previas, el primer diagnóstico diferencial era un daño miocárdico secundario de tipo isquémico por el cuadro de shock, pero no podían descartarse otras patologías miocárdicas primarias (p. ej una cardiomiopatía dilatada). Se instauró un tratamiento antiarrítmico con sotatol a una dosis baja de 1 mg/kg vía oral cada 12 horas por el efecto β negativo en un paciente con la función sistólica reducida.
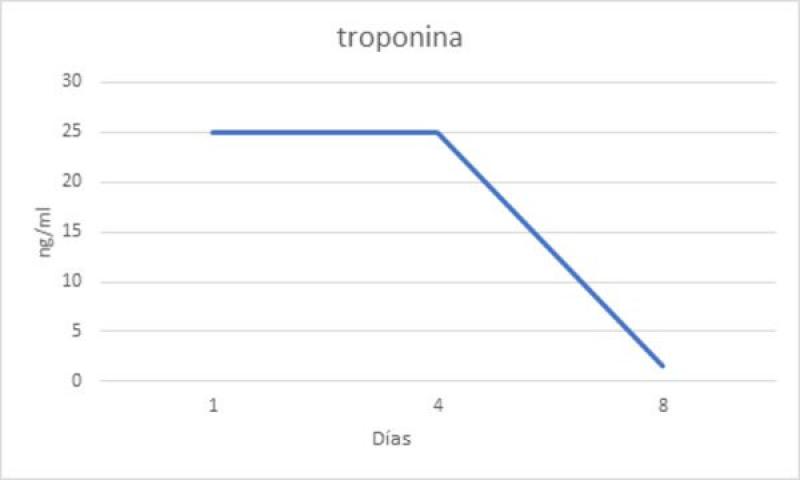
Se realizó una medición de la TnIc que reveló un valor muy elevado (>25 ng/ml) sugerente de un grave daño miocárdico agudo probablemente de tipo isquémico o inflamatorio. Al día siguiente el paciente mostró mejor actitud y una disminución notable del grado de dolor abdominal, por lo que se decidió reducir un 25% la velocidad de infusión continua de lidocaína-ketamina durante 6 horas hasta que finalmente se retiró. En este momento en la monitorización electrocardiográfica persistía un ritmo ventricular acelerado polimorfo, pero con una frecuencia ventricular menor respecto al día anterior. Se aumentó la dosis de sotalol a 2 mg/kg cada 12 horas y se añadió mexiletina 7,5 mg/kg cada 8 horas.
A pesar de que la paciente se encontraba clínicamente estable, presentó un episodio convulsivo tónico-clónico con pérdida de consciencia. Se administraron dos bolos de diazepam (0,5 mg/kg IV), pero no hubo respuesta. Se añadió levetiracetam a dosis de 20mg/kg IV y las crisis convulsivas se controlaron. El tiempo de recuperación fue de 4 horas hasta que el paciente mostró un estado mental alerta y recuperó tanto la repuesta a la amenaza como la sensibilidad consciente. Esta crisis convulsiva probablemente estuvo asociada a un accidente cerebro-vascular isquémico transitorio secundario a un estado de hipercoagulabilidad, daños de reperfusión, efectos adversos medicamentosos o patologías subyacentes intracraneales sin diagnosticar en el paciente.
El diagnóstico definitivo de la lesión requería la realización de pruebas de imagen avanzadas, así como de un perfil completo de coagulación, pero como el episodio fue aislado y la recuperación fue buena no fue necesario realizarlas. Se realizó una analítica sanguínea de control, evidenciando una mejoría considerable en los valores de creatinina sérica (de 2.4 mg/dl a su llegada al hospital a 1.1 mg/dl) tras recuperar el correcto estado de hidratación del paciente, sin embargo, se observó un aumento moderado de la ALT (160 U/L). El aumento de la ALT puede estar producido por una gran variedad de patologías (Tabla 5), pero probablemente en nuestro caso el aumento de la ALT era debido a una hipoperfusión hepática durante el shock hipovolémico.
| Tabla 5. Principales causas del aumento de las enzimas hapáticas en el perro. |
| Categorías | Intervalo de referencia |
| Metabólico | Lipidosis hepática, diabetes mellitus, amlloidosis, hiperadrenocorticismo canino (leve) |
| Neoplástico | Linfoma, carcinoma hepatocelular, metástasis, sarcoma histiocítica, etc. |
| Inflamatorio | Leptospirosis, hepatitis infecciosa canina, colangitis/colangiohepatitis, hepatitis crónica (idiopática, por cobre, por fármacos), cirrosis, pancreatitis, enfermedad intestinal inflamatoria |
| Hipóxico | Shock, pérdida aguda de sangre, insuficiencia cardíaca congestiva |
| Tóxico | Paracetamol, anestésicos, antiinflamatorios no esteroideos, antibióticos, antifúngicos, metimazol, etc. |
| Fármacos | Corticoesteroides, anticonvulsionantes |
| Miscelánea | Shunt portosistémico, migración hepática de parásitos, enfermedades de almacenamiento lisosomal |
| Músculo | Necrosis o trauma muscular, miopatía/miositis (sobre todo aumento de AST), distrofia muscular |
Al cuarto día de hospitalización el paciente estaba estable y la monitorización electrocardiográfica mostraba una reducción del número de arritmias. Predominaba un ritmo sinusal que se alternaba con salvas no sostenidas de ritmo idioventricular acelerado monomorfo con morfología de bloqueo de rama derecha (figura 2). La ecocardiografía mostraba una mejoría de los parámetros de función sistólica y los parámetros hemodinámicos eran normales. A pesar de la mejoría clínica y de los parámetros cardíacos, los valores de TnIc seguían elevados (>25 ng/ml). Los valores de TncI elevados asociados a la persistencia de arritmias ventriculares y la buena recuperación de la función cardíaca podían ser secundarios a una fase de reperfusión sin daños miocárdicos isquémicos graves (infartos). El paciente recibió el alta con sotalol 2mg/kg vía oral cada 12 horas y amoxicilina – ácido clavulánico 22mg/Kg vía oral cada 12 horas.
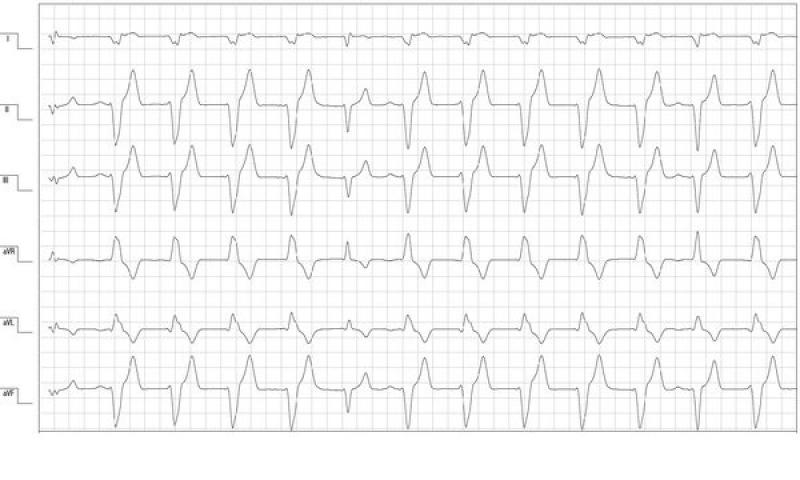
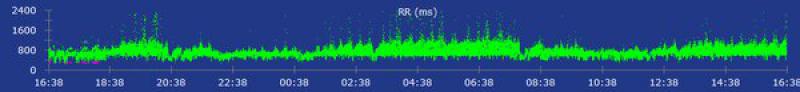
A los 4 días la paciente regresó de nuevo al centro para un seguimiento (8 días desde el momento de la hospitalización). Clínicamente estaba bien y el examen físico no mostraba alteraciones significativas. Se realizó una monitorización Holter de 24 horas en la que se observó un ritmo sinusal sin presencia de arritmias ventriculares y el examen ecocardiográfico no mostraba alteraciones funcionales (figura 3). En este momento la TnIc se había reducido significativamente (1,62 ng/ml). A las 4 semanas se realizó un Holter de revisión en el que se verificó la ausencia de arritmias y se pautó una reducción del fármaco antiarrítmico hasta suspenderlo completamente. En este momento no se obtuvieron valores de TnIc. A las 2 semanas se realizó un electrocardiograma de 5 minutos donde se observaba un ritmo sinusal sin presencia de arritmias.
La reducción de los valores de TnIc junto con la resolución del cuadro arrítmico y cardioestructural eran sugerentes de un daño miocárdico transitorio de tipo isquémico.
Discusión
En medicina humana, por definición, se considera daño miocárdico todas aquellas lesiones cardíacas en la que se produce una elevación de la Tnc por encima del límite superior de referencia del percentil 99 establecido en cada laboratorio9. Las causas de daño miocárdico se engloban en dos grandes grupos: isquémicas o no isquémicas (tabla 6)9. Se consideran lesiones isquémicas aquellas en la que el daño miocárdico ocurre como consecuencia de una anomalía en el aporte de oxígeno a la célula cardíaca (disminución/ interrupción de la perfusión o aumento de la demanda de oxígeno) y no isquémicas si la lesión miocárdica está provocada por otros mecanismos fisiopatológicos como por ejemplo la liberación de toxinas, alteraciones metabólicas o el daño traumático de la célula.
| Tabla 4. Número animales eliminaciones superiores e inferiores a 200hpg de strongilados. |
| Isquémicas: Isquemia miocárdica aguda producida por un desequilirio entre el aporte y la demanda de oxígeno | Disminución de la perfusión miocárdica |
|
| Isquémicas | Aumento de la demnada miocárdica de oxígeno |
|
| Reversible | Condiciones cardíacas |
|
| Reversible | Condiciones sistémicas |
|
La gravedad del daño miocárdico está relacionada con la causa que lo provoca y se considera mayor cuando el daño es de tipo isquémico. Las consecuencias de una lesión isquémica son variables y están relacionadas principalmente con la duración de la hipoperfusión, grado de la oclusión y la formación o no de un flujo colateral. Si la isquemia es prolongada y no se produce un flujo colateral a tiempo (en medicina humana más de 20 min si la oclusión es completa y más de 5 horas si la oclusión es parcial), el daño miocárdico se considera irreversible porque se produce la muerte (necrosis) de las células miocárdicas.
Esta situación se define como infarto miocárdico10. Si la oclusión es parcial o se crea un flujo colateral mínimo de mantenimiento el miocardio entra en una fase de disfunción transitoria (stunned myocardium o hibernating myocardium) en la que el tejido aún se considera viable y puede llegar a ser funcional una vez recuperado el flujo coronario (reperfusión)10.
Por último, si el flujo se restaura, pero ocurre en zonas de tejido donde el daño celular ha sido irreversible se producirán daños de reperfusión (zonas de hemorragia y necrosis coagulativa con bandas de contracción) que agravarán el daño miocárdico10. Desde el punto de vista clínico, en medicina humana el diagnóstico de infarto miocárdico es complejo y requiere la evidencia de necrosis miocárdica a través de la identificación de una serie de criterios clínicos: síntomas, alteraciones hematológicas y anomalías electrocardiográficas o en pruebas de imagen9.
Como hemos introducido al principio del artículo, la Tnc es uno de los principales marcadores biológicos específicos de daño miocárdico, pero existen otras moléculas que pueden ser utilizadas como marcadores. Cuando la membrana celular del cardiomiocito se daña en una situación de isquemia/necrosis, se liberan a la circulación gran cantidad de sustancias disueltas en el citoplasma y otras macromoléculas (enzimas, proteínas estructurales, etc).
Entre las moléculas que se liberan con mayor facilidad, se encuentran aquellas sustancias que se localizan en el citoplasma, como pueden ser iones y metabolitos (lactato), los cuales no se consideran marcadores específicos cardíacos ya que presentan una distribución tisular muy amplia (figura 4)11. Otra macromolécula de naturaleza enzimática que se utiliza como marcador biológico de necrosis miocárdica es la creatina quinasa total (CK), más concretamente su isoenzima cardíaca (MB), pero no es específica de daño miocárdico si además coexiste un daño musculoesquelético ya que se encuentra tanto en el músculo esquelético como en el miocardio.
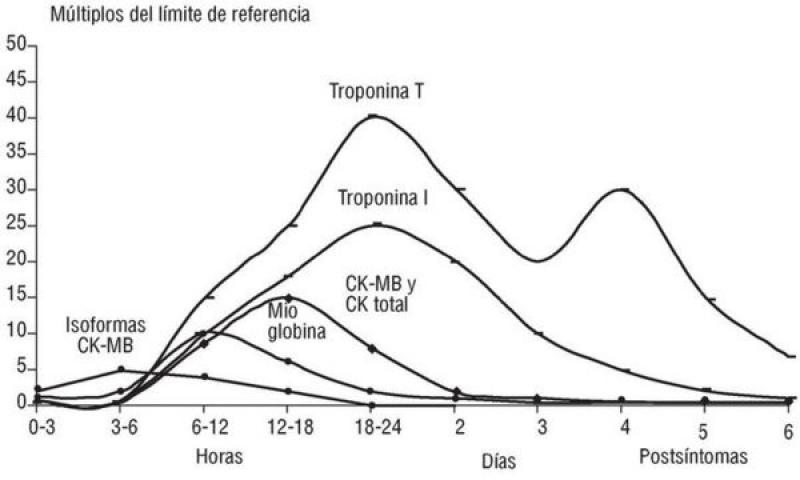
En caso de infarto miocárdico la actividad/concentración de CK-MB aumenta a partir de la 4-6 horas del inicio del daño isquémico, permanece elevada hasta 24-36 horas y luego desciende rápidamente. La mioglobina es una proteína citoplasmática que se libera de forma muy rápida (1-2 horas), alcanza su concentración máxima en plasma entre 6-12 horas y desaparece en 12-24 horas (figura 4)11. La mioglobina no resulta muy útil como marcador biológico porque no es cardioespecífica y su detección en plasma debe ser muy precoz. A diferencia de la CK y la mioglobina, la troponina posee isoformas específicas del músculo cardíaco (TnTc y TnIc).
La cinética de liberación de la TnTc y la TnIc es diferente. La TnTc tiene un valor máximo inicial a las 12 horas, seguida de una meseta hasta las 48 horas y una fase de descenso con una duración variable (7-21 días). La TnIc presenta una cinética similar, pero con valores ligeramente inferiores y una fase de descenso más breve (figura 4)11. A pesar de que en nuestro caso no hemos obtenido un valor de TnIc el primer día, y que no ha sido posible realizar mediciones seriadas de la TnIc, los valores de TnIc siguen un patrón similar al descrito en medicina humana (figura 5). Los valores superaban los 25 ng/ml en la primera medición, mantuvieron una fase de meseta aproximadamente durante 3 días y luego descendieron hasta alcanzar casi la normalidad 8 días después de identificar el daño miocárdico.
La isquemia miocárdica provoca cambios significativos en la célula miocárdica, tanto intracelulares como extracelulares12. Entre los cambios celulares se incluyen las anomalías metabólicas (acidosis), alteraciones iónicas (aumento de las concentraciones de iones como el potasio, sodio y magnesio) o la liberación de catecolaminas12. Estos cambios bioquímicos y metabólicos alteran las corrientes iónicas a través de la membrana del cardiomiocito provocando alteraciones en el potencial de acción: anomalías en la despolarización, reducción de la velocidad de conducción, disminución de la excitabilidad, reducción del potencial de acción, alteraciones en la repolarización, dispersión de la repolarización o automatismo anormal12.
Todos estos cambios determinan unas alteraciones en el electrocardiograma de superficie y sirven como substrato arrítmico para el desarrollo de diferentes arritmias (taquicardias y bradiarritmias) o trastornos de la conducción12. A pesar de que la gran mayoría de los estudios acerca de los mecanismos de las arritmias en la isquemia e infarto miocárdico han sido realizados en modelos animales, en medicina veterinaria no existe una definición clara de los cambios electrocardiográficos en caso de cardiomiopatía isquémica. Las alteraciones electrocardiográficas sugerentes de daño isquémico suelen ser anomalías en la repolarización (cambios en el segmento ST o en la onda T)13.
En medicina humana existen criterios electrocardiográficos más específicos para el diagnóstico de infarto miocárdico6. En nuestro caso el paciente presentó arritmias ventriculares que variaron en número, morfología y organización a lo largo del tiempo. Durante isquemia e infarto miocárdico las arritmias ventriculares pueden aparecer en distintas fases y en cada una de ella presentan unas características diferentes12. La fase aguda ocurre aproximadamente 20-30 minutos después de la oclusión de la arteria coronaria y es cuando se considera que los cambios son reversibles. La mayor parte de esta fase se caracteriza por una acumulación local elevada de catecolaminas y por un aumento de la automaticidad en las células.
La segunda fase ocurre 72 horas después de la oclusión del vaso, con un pico entre las 12-24 horas. En esta fase las arritmias están provocadas principalmente por un automatismo anormal o actividad desencadenada dentro de las fibras de Purkinje que aún estén intactas, y con menos frecuencia por mecanismos de reentrada. La última fase o fase crónica se desarrolla transcurridas 72 horas y normalmente las arritmias ventriculares son consecuencia de circuitos de reentrada creados por la presencia de zonas de infarto.
En nuestro caso, el cuadro arrítmico comenzó con la presencia de escasos complejos ventriculares polifocales que progresaron en 24 horas a un ritmo idioventricular acelerado polimorfo alternado con períodos de taquicardia ventricular monomorfa con morfología de bloqueo de rama izquierda. El mecanismo responsable de la mayoría de las taquicardias ventriculares monomorfas suelen ser los circuitos de reentrada, mientras que los ritmos ventriculares polimorfos pueden estar provocados tanto por un mecanismo de reentrada como por actividad desencadenada13.
Estas arritmias ventriculares pueden estar presentes tanto en patologías cardiacas estructurales (primarias o secundarias) como en ausencia de cardiopatía, pero en nuestro caso las arritmias eran sugerentes de un daño miocárdico isquémico en fase aguda por las características electrocardiográficas y porque los valores de TnIc eran muy elevados. A los 4 días el cuadro arrítmico cambió y sólo se observaban salvas de ritmo idioventricular acelerado monomorfo. En este momento los valores de troponina aún estaban elevados. Probablemente el paciente se encontraba en una fase crónica con algún cambio isquémico más grave que favorecía la formación de circuitos de reentrada (zonas de reperfusión). Finalmente, transcurrida una semana del inicio de los síntomas, no se identificaron arritmias durante el Holter. La ausencia de arritmias y un valor normal de la TnIc confirmaban la resolución de daño miocárdico agudo.
Los pacientes con daño miocárdico suelen presentar un grado variable de disfunción y remodelación cardíaca. Los cambios están asociados a la gravedad y extensión de la lesión, y de estos factores también dependerá que el daño sea reversible o no10. En el caso de patologías isquémicas el grado de disfunción miocárdica está principalmente influenciado por la proporción de áreas de infarto y la cantidad de miocardio no infartada o miocardio viable que se recupera tras la reperfusión10. Tanto la función sistólica como diastólica del ventrículo se ven alteradas y como consecuencia suele producirse un aumento de las cámaras cardíacas y signos de bajo gasto o insuficiencia cardíaca congestiva10.
En nuestro caso no disponemos de parámetros ecográficos u otras técnicas diagnósticas más específicas para valorar la función cardíaca, pero los parámetros ecocardiográficos estándar nos permitieron intuir la presencia de una hipocinesia difusa. La función sistólica se recuperó completamente una vez normalizados los valores de troponina y la desaparición de las arritmias ventriculares, por lo que es probable que se haya tratado de una disfunción cardíaca transitoria ((stunned myocardium o hibernating myocardium). Parte de la disfunción miocárdica probablemente era secundaria al daño isquémico, pero la presencia de las arritmias también pudo contribuir a reducir la función cardíaca.
Los mecanismos que contribuyen al daño miocárdico en curso de enfermedades sistémicas son múltiples: hipotensión, hipoxemia, anemia, fiebre, taquicardia, estrés miocárdico, tromboembolismo, microtrombos, efectos tóxicos de las endotoxinas y citoquinas, estrés oxidativo, daños de reperfusión2. En nuestro caso, se describe una situación de shock hipovolémico como consecuencia de la hemorragia intraabdominal aguda ocasionada por la rotura de una masa esplénica. En estas situaciones se produce una grave reducción del volumen sanguíneo intravascular, y por tanto una grave hipoperfusión tisular sistémica14.
En estados de shock hipovolémico, la reducción del gasto cardíaco ocasionada por la disminución del retorno venoso desencadena una serie de mecanismos compensatorios, cuyo objetivo es mantener la presión arterial sistémica, pero que en algunos casos incluso pueden ser perjudiciales si la respuesta se mantiene por un largo periodo de tiempo14. Uno de los principales mecanismos de compensación es el aumento de la actividad simpática que provoca vasoconstricción, aumento de la contractilidad y frecuencia cardíaca14. Esta respuesta aumenta el gasto cardíaco, pero en detrimento también incrementa el consumo de oxígeno en el miocardio14. El desequilibrio entre la demanda metabólica de oxígeno y el oxígeno transportado a los tejidos se traduce en un déficit de oxígeno en los tejidos que conlleva un metabolismo celular anaerobio con aumento de la producción de lactato, acidosis metabólica y cambios celulares15.
Como hemos visto la cardiopatía isquémica o daño miocárdico isquémico es una entidad ampliamente descrita en medicina humana, pero por lo contrario poco conocida en medicina veterinaria (principalmente sólo en estudios postmortem o modelos experimentales)16. Este hecho probablemente está relacionado con la escasa predisposición de los perros y los gatos a patologías coronarias (arterioesclerosis, ateroesclerosis, etc.), a que poseen una circulación coronaria colateral muy bien desarrollada que probablemente les protege de la isquemia o a que sea, posiblemente en muchas ocasiones, una patología infradiagnosticada17. El uso de marcadores biológicos como la TnIc en casos en los que la sospecha clínica sea un daño miocárdico agudo es muy útil para definir la etiología del proceso, monitorizar la progresión de la enfermedad y dar un valor pronóstico.
Bibliografía
- Langhorn R, Willesen JL. Cardiac troponins in dog and cats. J Vet Intern Med. 2016; 30:36-50.
- Oyama MA. Using cardiac biomarkers in veterinary practice. Vet Clin North Am Small Anim Pract 2013;43:1261-1272.
- Katz AM. The contractile proteins. In: Physiology of the heart, ed 5ª, Philadelphia; 2011: 88-107.
- White DH. Pathobiology of troponin elevations: do elevations occur with myocardial ischemia as well as necrosis?. J Am Coll Cardiol 2011; 14: 2406-2408.
- Agewall S, Giannitsis E, Jernberg T, Katus H. Troponin elevation in coronary vs non-coronary disease. Eur Heart J 2011;32:404-411.
- Porter A, Rozanski E, Price LL, Shaw S. Evaluation of cardiac troponin I in dogs presenting to the emergency room using a point-of-care assay. Can Vet J 2016; 57: 641-645.
- Langhorn R, Oyama MA, King LG, Machen MC, Trafny DJ, Thawley V, Willesen JL Tarnow I, Kjelgaard-Hansen. Prognostic importance of myocardial injury in critically ill dogs with systemic inflammation. J Vet Intern Med 2013; 27:895-903.
- Hamacher L, Dörfelt R, Müller M, Wess G. Serum cardiac troponin I concentrations in dogs with systemic inflammatory response syndrome. J Vet Intern Med 2015; 29:164-170.
- Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, White HD; Executive Group on behalf of the Joint European Society of Cardiology (ESC)/American College of Cardiology (ACC)/American Heart Association (AHA)/World Heart Federation (WHF) Task Force for the Universal Definition of Myocardial Infarction. Fourth Universal Definition of Myocardial Infarction (2018). J Am Coll Cardiol. 2018; 30:2231-2264.
- Canty JM, Duncker DJ. Coronary blood flow and myocardial ischemia. In: Braunwald’s Heart Disease. A textbook of cardiovascular medicine. Elsevier Saunders, Philadelphia. 2015. Tenth edition.
- Santaló M, Guindo J, Ordóñez J. Marcadores biológicos de necrosis miocárdica. Revista española de cardiología. 2003; 56:703-720.
- Ghuran AV, Camm AJ. Ischaemic heart disease presenting as arrhythmias. British Medical Bulletin 2001;59:193-210.
- Santilli R, Möise NS, Pariaut R, Perego M. Electrocardiography of the dog and cat. 2nd edition. Edra, Milan. 2018.
- Hall, JE. (2020). Circulatory shock. En: Guyton and Hall Textbook of Medical Physiology (12th ed., pp. 273-282). Elsevier.
- Bounos G, Hampson LG, Gurd FN (1963). Regional blood flow and oxygen consumption in experimental hemorrhagic shock. Arch surg, 87:340-54.
- Kidd L, Stepien RL, Amrheiw DP. Clinical findings and coronary artery disease in dogs and cats with acute and subacute myocardial necrosis: 28 cases. J Am Anim Hosp Assoc 2000;36:199–208.
- Verdouw PD, van den Doel MA, de Zeeuw S, Duncker DJ. Animal models in the study of myocardial ischaemia and ischaemic syndromes. Cardiovasc Res 1998;39:121–135.